Epigenetic Modifiers
Epigenetic Modifiers in Disease and Development
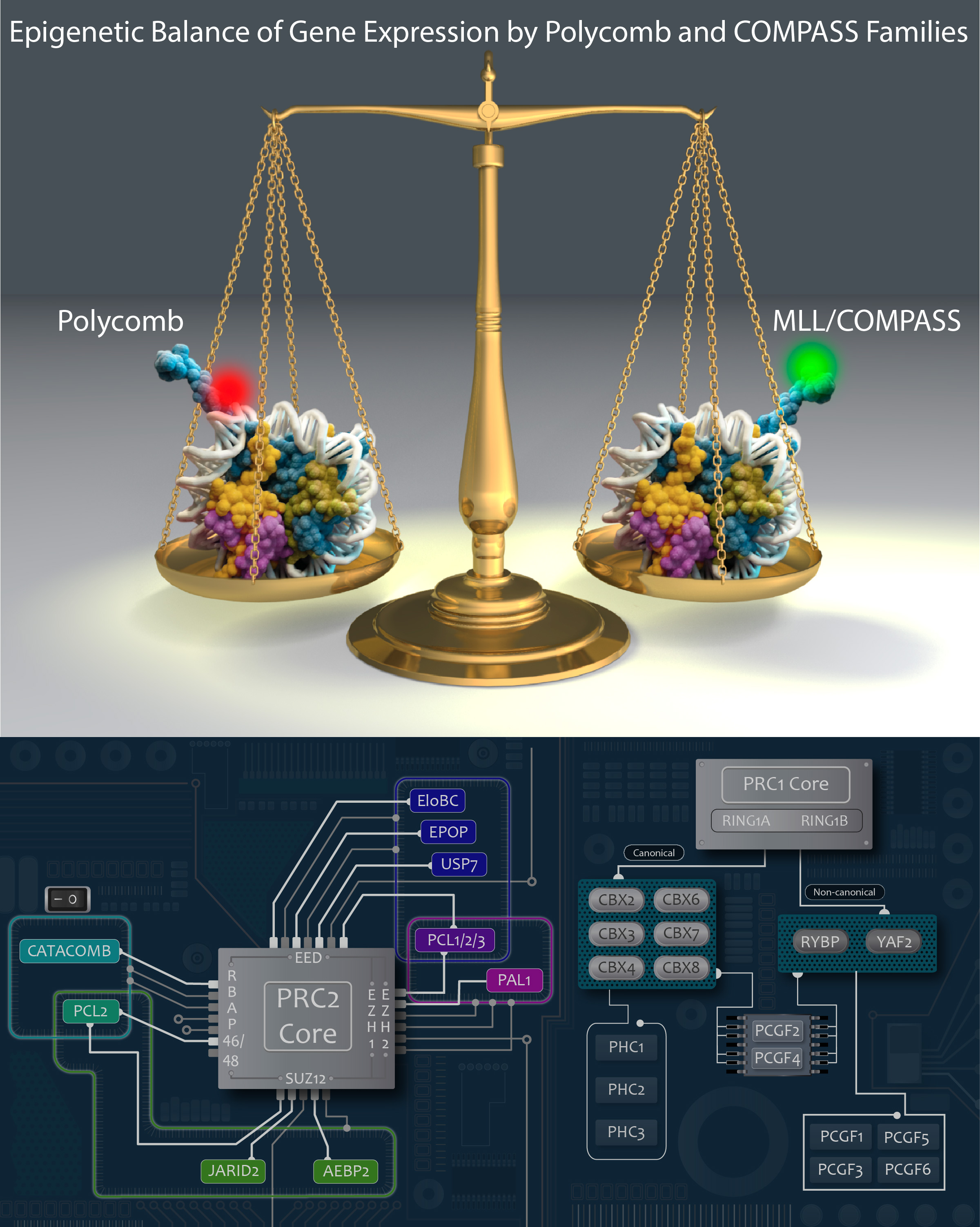
After identifying and characterizing COMPASS in yeast, our work in Drosophila further elucidated the Set1/Trx/COMPASS family of histone-modifying methyltransferase complexes, which are evolutionarily conserved. Extending our understanding to mammalian systems we demonstrated that, like its Drosophila homolog Trithorax (Trx), mammalian MLL1 similarly marks CpG-dense regions similar to Polycomb response elements (PREs) with H3K4me2. Importantly, we also showed that this conserved activity exists in a functional balance with that of the Polycomb repressive complex (PRC2) and its H3K27me3 mark. [Rickels 2016]
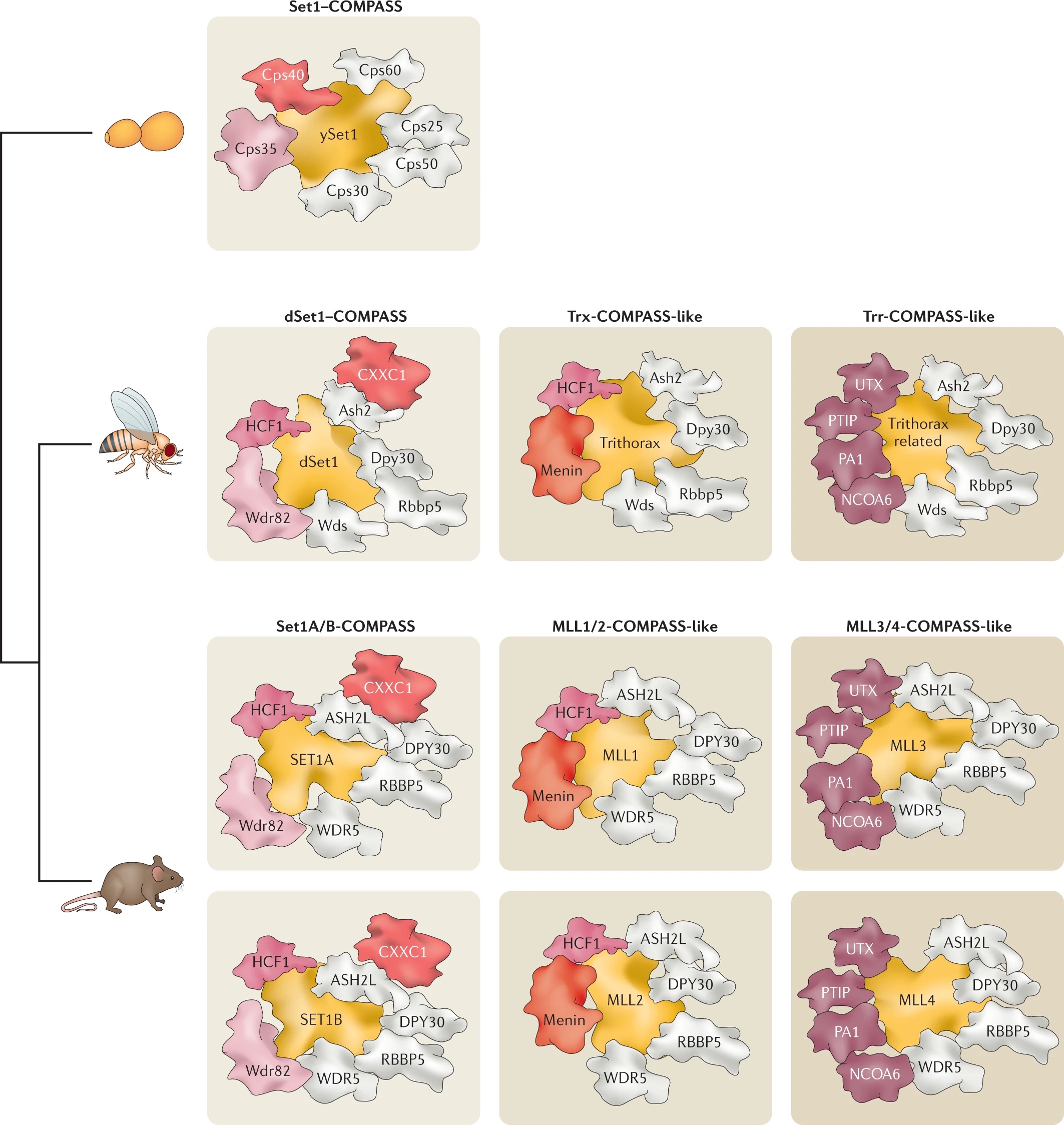
Now we work exclusively in mammalian systems, where the mechanisms of epigenetic modification are somewhat more complex. Expanding upon the one in yeast and three in flies, mammals have a total of six COMPASS methyltransferase family members, in three “branches”: Set1A/B, MLL1/2, and MLL3/4. Though any of these enzymes can associate with scaffolding and accessory proteins to form a COMPASS complex, the resultant complexes are distinct, with different protein constituents and regulatory functions (read more in our recent review: [Cenik 2021]). For example, we found that the activation of early Hox genes in response to the nuclear receptor agonist retinoic acid specifically requires Set1A, but not MLL1-4, in murine embryonic stem cells (mESCs, in which Set1B is not expressed.) [Cao 2017]
Mammalian development and differentiation also require DNA methylation, a process of repressive chromatin modification that appears to be absent in Drosophila. The crosstalk between DNA methylation and histone modification may play a role in how mammalian cells respond to specific stimuli. For example, we discovered a positive feedback loop in which activation of the nuclear estrogen receptor ERa induces expression of the methylcytosine dioxygenase TET2, which in turn initiates DNA demethylation at the enhancers of ERa target genes. We found that ERa induces TET2 expression by binding to enhancer elements upstream of the TET2 promoter, and further determined that the presence of MLL3, but not the closely related MLL4, is specifically required for this binding. [Wang 2018]
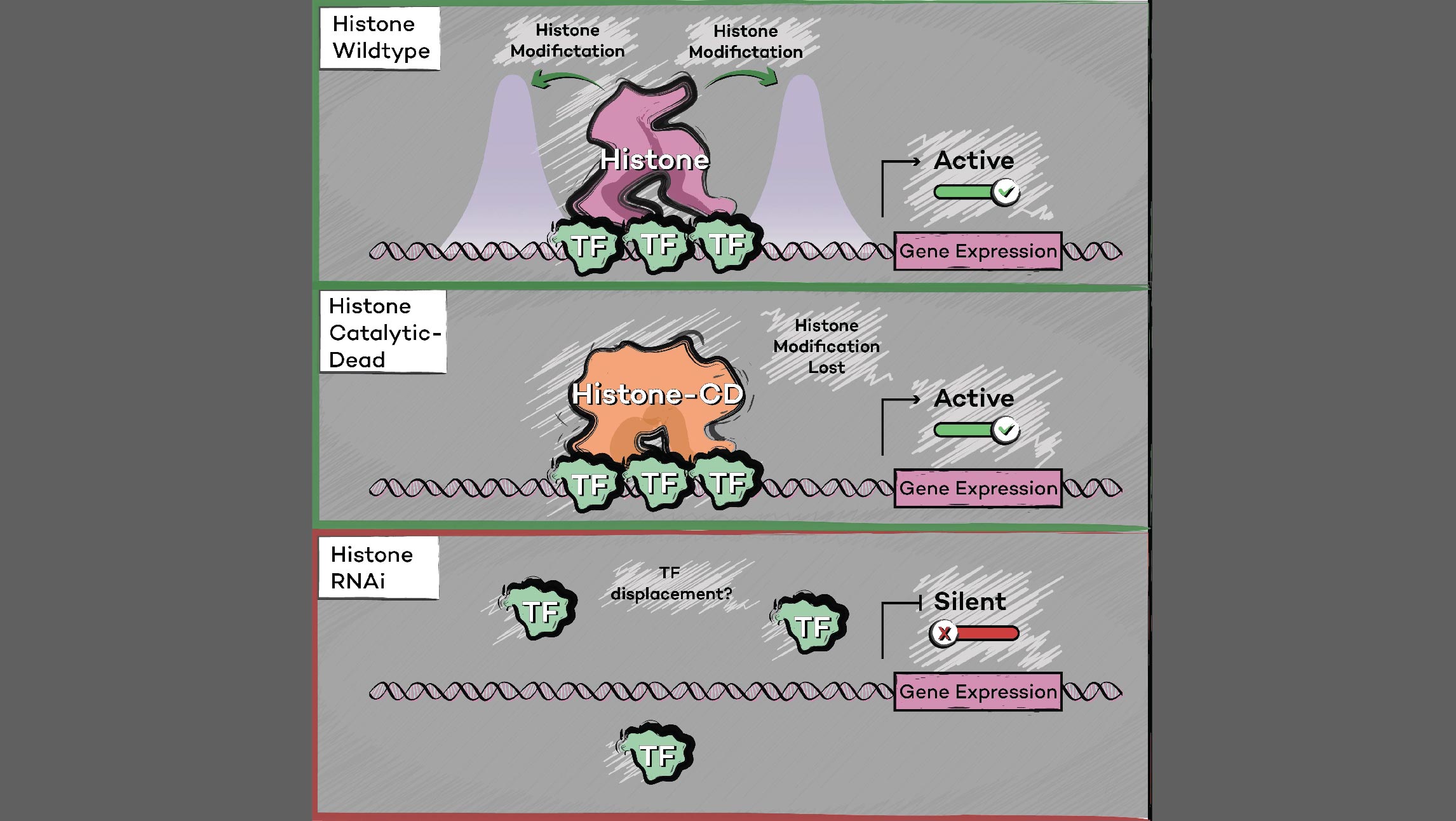
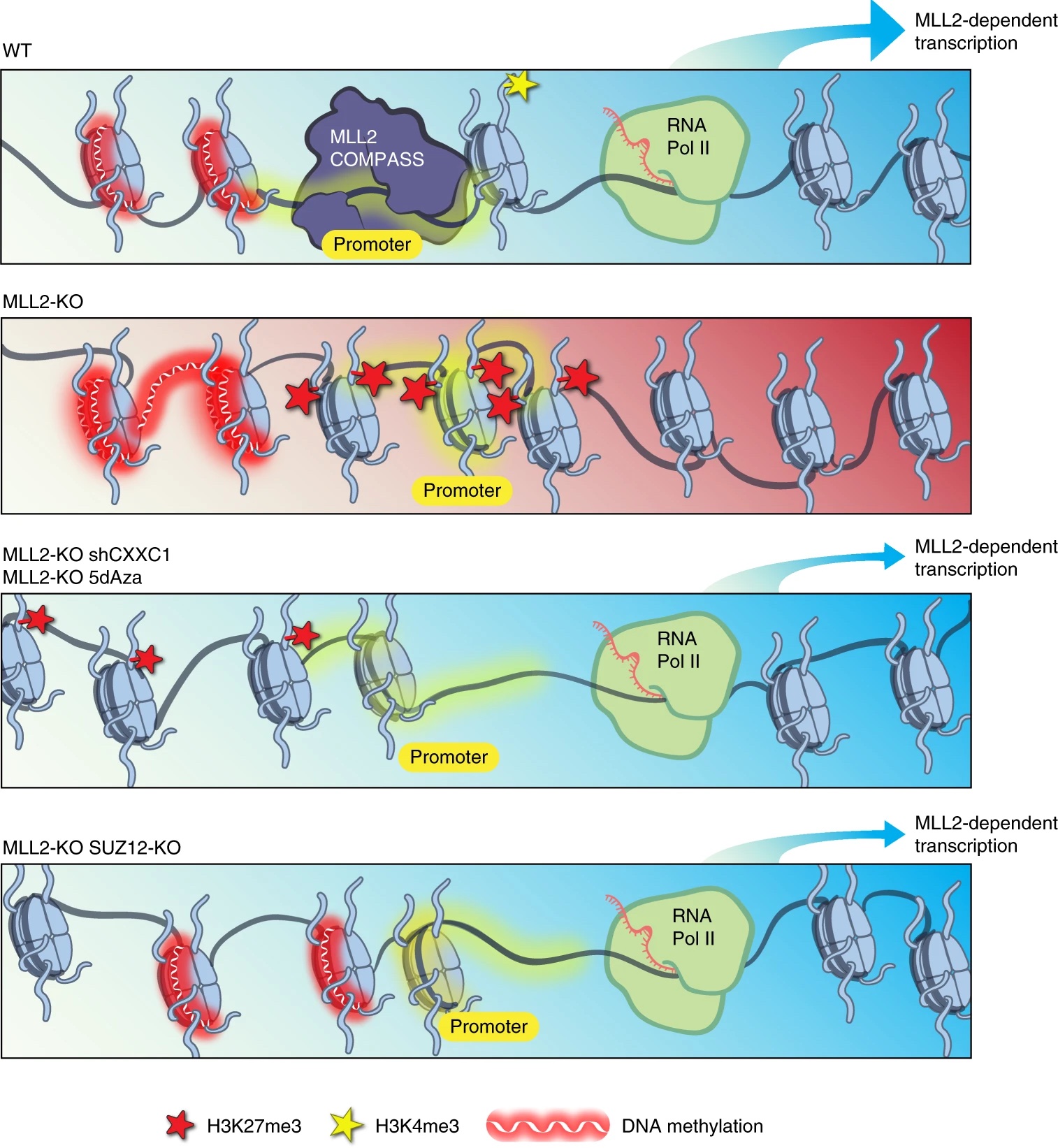
We recently identified a role for DNA methylation in the functional balance between COMPASS and Polycomb. The results of a CRISPR screen for factors regulating MLL2-dependent gene expression indicated a surprising balance between COMPASS family members, in which the absence of MLL2 allows for inhibition mediated by Set1A/B. We further demonstrated that knockdown of the Set1A/B component CXXC1 could rescue MLL2-dependent gene expression, and that it did so without restoring H3K4me3. Expression of the DNA methyltransferase DNMT1 is reduced in CXXC1 KO mESCs, and we found that either DNMT1 KD or global demethylation with the compound 5dAza could rescue gene expression in MLL2 KO mESCs. However, the vast majority of MLL2-dependent gene promoters had minimal CpG methylation unchanged by these knockdown or inhibition conditions, indicating that the repressive effect of DNA methylation on MLL2-dependent genes must be indirect. We guessed that Polycomb might be a third player, and indeed found not only that CXXC1 KD could rescue an increase in H3K27me3 at MLL2-dependent gene promoters but also that knockout of the Polycomb component SUZ12 could rescue MLL2-dependent gene expression in MLL2 KO mESCs. [Douillet 2020]
Many human cancers are driven by translocations and other mutations that affect epigenetic modifiers. In MLL, stability of the translocation-generated chimera is often dramatically enhanced relative to the wild type allele. Investigating mechanisms of MLL1 destabilization, we found that wild type but not chimeric MLL1 is destabilized both by UBE20-mediated degradation downstream of IL-1 signaling and by Taspase 1 cleavage, which is facilitated by casein kinase II (CK II) phosphorylation proximal to the cleavage site. We were able to stabilize MLL1 by blocking these degradation and cleavage pathways with IRAK and CKII inhibitors, respectively. Once stabilized, wild type MLL1 was able to displace chimeric MLL1 from target genes to relieve oncogenic “addiction.” We demonstrated delayed leukemic progression and increased survival after treatment with either inhibitor in a murine MLL-AFF9 model, further identifying IRAK or CKII inhibitors as potential therapeutics in MLL-rearranged leukemias. [Liang 2017, Zhao 2019]
Polycomb translocations also generate oncogenic fusion chimeras. We recently identified a previously uncharacterized PRC2 subunit in an endometrial stromal sarcoma-associated fusion protein; we termed this subunit CATACOMB (catalytic antagonist of Polycomb), because we found that it decreases PRC2-mediated H3K27 methylation. Further dissecting CATACOMB’s inhibitory function, we identified a critical methionine within a sequence strikingly similar to that of histone tails containing a H3K27M substitution. [Piunti 2019] This substitution is present due to point mutation in more than 80% of diffuse intrinsic pediatric glioma (DIPG) tumors, and it decreases PRC2-mediated H3K27 methylation. Our epigenetic profiling of H3K27M DIPG tumor cells revealed an increase in both the H3K27ac mark and its recognizant bromodomain proteins, which exclude PRC2 from actively transcribed genes. We further demonstrated that BET inhibitors could block bromodomain protein recruitment to heterotypic H3K27M-H3K27ac nucleosomes and effectively slow DIPG tumor progression, therefore identifying BET bromodomain inhibitors as potential therapeutics for DIPG. [Piunti 2017] H3K27 methylation can also be decreased by exogenous inhibition of the PRC2 catalytic subunit EZH2, which is elevated in advanced urothelial carcinoma (UCa). We recently demonstrated that small molecule EZH2 inhibition reduced tumor progression in a carcinogen-induced bladder cancer model by increasing immune cell infiltration and activating adaptive immunity. [Piunti et al., 2022]
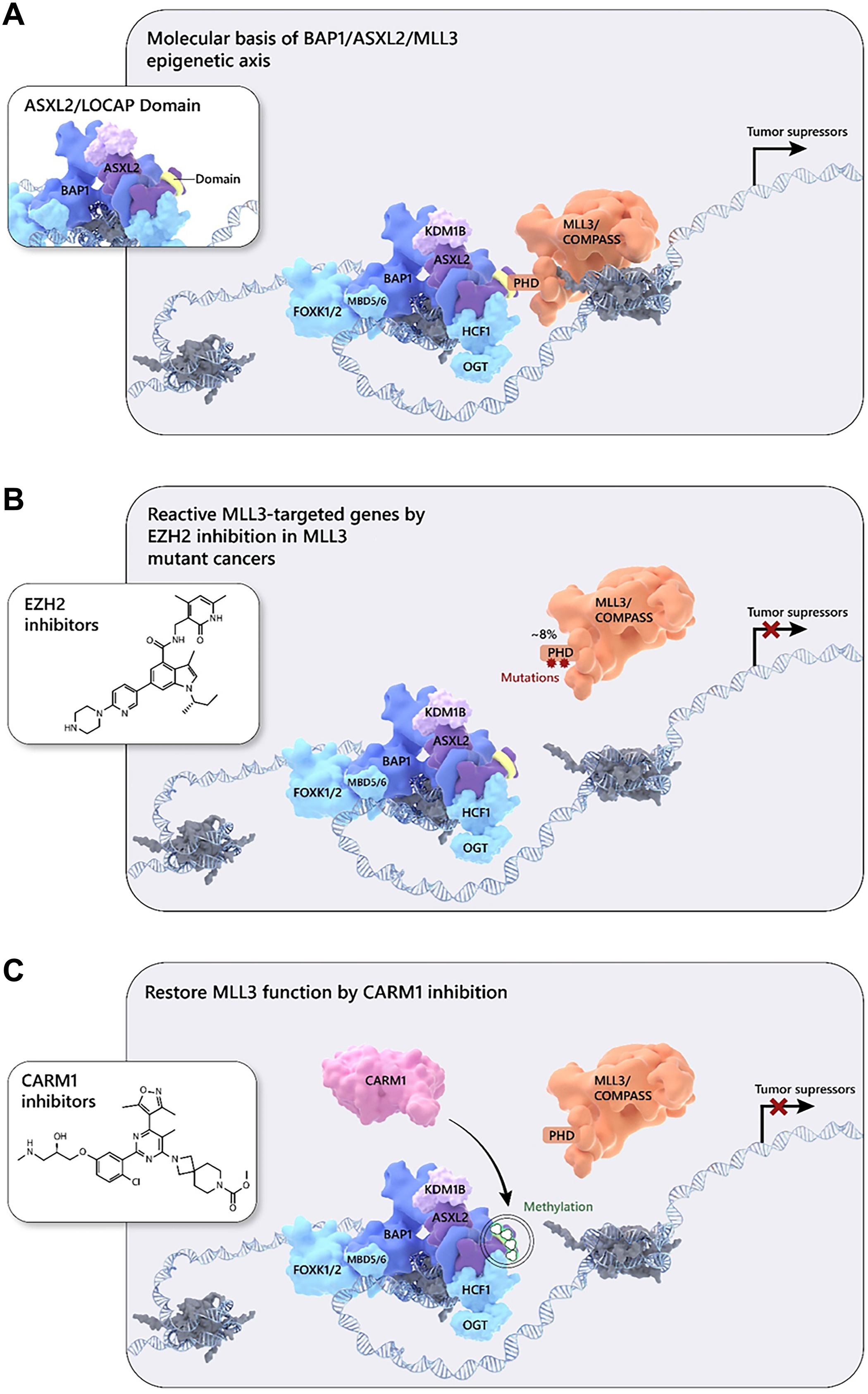
The balanced opposition between COMPASS- and Polycomb-mediated histone modification is essential to development, and its dysregulation results in cancer and other disease. However, their relationship presents an opportunity: disease arising from COMPASS dysfunction can be addressed via Polycomb targeting, and vice versa. For example, we found that mutations within the plant homeodomain (PHD) repeats of MLL3 disrupt its interaction with the H2A deubiquitinase BAP1, reducing recruitment of MLL3 and its H3K27 demethylase partner UTX to enhancers. However, “resetting” the epigenetic balance at enhancers by inhibiting PRC2 restored normal gene expression in cancer cells harboring BAP1 or MLL3 mutations. [Wang 2018] What if, instead, you wanted to inhibit MLL3 recruitment to restore a balance perturbed by PRC2 dysfunction? In our follow up study, we determined that it is the BAP1 subunit ASXL2 specifically that recruits MLL3 to its target loci, and that it does do via a direct interaction with MLL3, which can be blocked by activity of protein arginine methyltransferase PRMT4/CARM1. This study identified a new target for the COMPASS side of the epigenetic balance at enhancers. [Zhao 2022]
Active Areas of Investigation